Страница
11
- Общий белок и белковые фракции1
- Трансаминазы АсАТ, АлАТ1
- Маркеры вирусов гепатита В и С в крови1
- С-реактивный белок1
- Сывороточные иммуноглобулины А, М, G1
- Субпопуляции лимфоцитов2
- Концентрация общего IgE в сыворотке1
2. Дополнительные лабораторные исследования:
- Определение уровня паратгормона в плазме1
- Вирусные маркеры (антитела к вирусу Эпштейна-Барр)1
3. Обязательные инструментальные исследования:
Рентгенография органов грудной клетки
УЗИ сердца
ЭКГ
4. Дополнительные инструментальные исследования:
УЗИ органов брюшной полости - 1.
5. Консультации специалистов (по показаниям):
Эндокринолога - 1;
Кардиохирурга - 1;
Отоларинголога - 1;
Окулиста - 1. [7]
Характеристика лечебных мероприятий: иммунная недостаточность, за редким исключением, не определяет прогноз и спектр ведущих клинических проявлений при СДД. В большинстве случаев, если пациент переживает 6-месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета.
Коррекция Т-клеточных нарушений при СДД может быть достигнута трансплантацией фетального тимуса. При наличии тяжелых пороков, в основном определяющих прогноз для жизни, пересадка тимуса считается недостаточно обоснованной. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез - по стандартам эндокринологических отделений [9].
Для лечения синдрома Ди Джорджи с успехом применяют пересадку костного мозга. Кроме того, достигнуты положительные результаты при пересадке эмбрионального тимуса больным с тяжелой степенью поражения [11].
Лечение: трансплантация или имплантация ткани тимуса. Введение тимозина (экспериментально). Противосудорожная терапия, введение препаратов кальция [11].
Длительность стационарного лечения определяется характером и тяжестью клинических проявлений и инфекционных осложнений и варьирует от 3 недель до 3 месяцев [9].
Требования к результатам лечения - купирование клинических проявлений (осложнений) СДД [9, 10].
ВЫВОДЫ
В представленной работе были рассмотрены микроделяционные синдромы: Прадера-Вилли, Ангнльма и Ди Джорджи. Описаны их клинические и генетические проявления, возможные риски. И представлены медицинские методы купирования их проявлений, улучшения состояния больных.
На сегодняшний день синдромы Прадера-Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений - геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера-Вилли может быть обусловлен двумя основными механизмами. Первый из них - микроделеция хромосомы 15 (15q11.2-q13), которая всегда отцовского происхождения. Второй - материнская изодисомия, т.е. когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера - Вилли обусловлено микроделецией, остальные - дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Для синдрома Ди Джорджи установлено, что он вызывается аутосомным генетическим дефектом делеции 22q11.2. Цитогенетические проявления данного синдрома на сегодня еще не достаточно изучены. Учеными ведется активная работа по решению этой проблемы.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Гинтер Е.К. Медицинская генетика: Учебник. – М.: Медицина, 2003. – 448с.
2. Ридли М. Геном. Автобиография вида в 23 главах. – М.: Эксмо, 2008. – 432с.
3. Денисенкова Е.В., Петрин А.Н., Байков А.Д. Цитогенетические и молекулярно-цитогенетические аспекты диагностики заболеваний, сопровождающихся умственной отсталостью, у детей // Издательство «Медиа Сфера», 2005. Режим доступа:
http://www.mediasphera.ru/journals/detail/206/2988/
4. Сапиенца К. Геномный импринтинг // В мире науки. (Scientific American. Издание на русском языке). - 1990. - №12. - стр.14-20. Режим доступа: http://www.evolbiol.ru/gen.html
5. Дуліпа О.Г. Синдром Прадера-Віллі (Prader-Willi Syndrome). Режим доступа: http://www.ibis-birthdefects.org/start/ . prader.htm
6. Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В. Московский НИИ педиатрии и детской хирургии Минздрава РФ. Синдром Прадера - Вилли у детей: новое в этиологии, патогенезе и лечении. Режим доступа: http://nature.web.ru/db/msg.html?mid=1174772&uri=index2.html
7. Миронов М.Б., Мухин К.Ю., Кузина Н.Ю., Боровиков К.С., Гоева И.А., Красильщикова Т.М., Коркин А.В., Петрухин А.С. Синдром Ангельмана. Клинический случай. Режим доступа: http://www.epileptologist.ru/pub/Angelman.html
8. Белозеров Ю.В., Леонтьева И.В., Школьникова М. А., Страхова О.С., Себелева И.А., Макаров Л.М., Давыдкин В.В., Динов Б.А. Наследственные болезни сердца у детей // Мир науки и культуры, 2000-2009. Режим доступа: http://nature.web.ru/db/msg.html?mid=1166264&s=111400060
9. Соколов В.Н. Гипопаратериоз // Наука и практика, 2009. Режим доступа: http://www.chtfoms.ru/index.php?option=com_content&task=view&id=26467&Itemid=24
10. Ярцев М.Н., Яковлева К.П., Плахтиенко М.В. ГНЦ Института иммунологии ФМБА России, Москва Иммунодефицитные состояния у детей // Издательство Media Medica, 2000. Режим доступа: http://old.consilium-medicum.com/media/pediatr/06_01/4.shtml
11. Вилочковой железы аплазия врожденная (Ди Джорджа синдром). Режим доступа: http://nasledbolezn.org.ua/obmen-veshestv/didjordja-sindrom.html
ПРИЛОЖЕНИЕ

Рис. 1. 15 хромосома


Рис. 2. Делеция 15 q11 – 13.1

Рис. 3. Дети с синдромом Прадера-Вилли

Рис. 4. Мальчик с синдромом Прадера-Вилли

Рис. 5. Синдромы Прадера-Вилли и Ангельмана

Рис. 6. Мальчик с синдромом Ангельмана
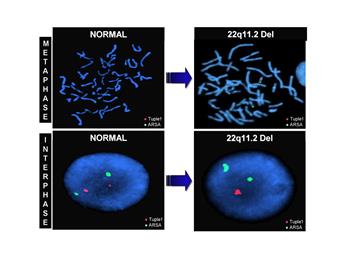
Рис. 7. Делеция 22q11.2 – синдром Ди Джорджи