Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи
Если все анализы дали отрицательный результат, необходимо провести полное цитогенетическое и молекулярно-генетическое исследования региона 15q11-q13, исключить дефицит метилен-тетрагидрофолат-редуктазы и мутации в гене АТR-Х [6]. Те же авторы приводят алгоритм обследования больных с мышечной гипотонией и Прадер-Вилли-подобным фенотипом. Этим пациентам необходимо провести ДНК-исследование районов метилирования, исключить синдром FRA-Х, затем осуществить ДНК- анализ на однородительскую дисомию хромосомы 14 и в случае отрицательных результатов провести исследование для выявления МЕСР2 мутаций. Таким образом, чтобы провести полный генетический анализ при подозрении на синдромы Ангельмана или Прадера-Вилли, в некоторых случаях необходимо применить до 8 различных цитогенетических, молекулярно-цитогенетических, молекулярных методов. Пациенты с подозрением на указанные синдромы являются примером многостороннего и целенаправленного исследования, необходимого для правильной постановки диагноза больным [6].
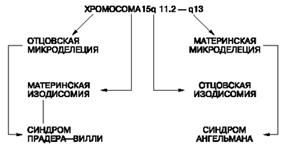
Рис. 5. Возможные механизмы развития синдромов Прадера-Вилли и Ангельмана
В заключение следует еще раз подчеркнуть, что более широкое комплексное применение цитогенетических, молекулярно-цитогенетических методов лабораторного анализа в сочетании с грамотным клиническим обследованием позволит решить проблему диагностики изолированной умственной отсталости или умственной отсталости при наследственных болезнях и синдромах у детей. Это в свою очередь поможет более эффективно осуществлять помощь членам наследственно отягощенной семьи в решении вопросов прогноза потомства и снизить частоту рождения детей с генетическими патологиями.
ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА)
Синдром Ди Джорджи (Ди Георга, СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. Впервые описан в 1965 году. Поражаются чаще девочки [9, 10, 11].
Наследуется по аутосомно - рецессивному типу [11]. Установлена генетическая характеристика этого врожденного порока развития, в части случаев это аутосомный генетический дефект делеции 22q11.2.
В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты [9].
Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания [9].
Тимус и паращитовидные железы образуются из третьего и частично четвертого карманов. Паращитовидные железы участвуют в регуляции уровня кальция в крови. Отсутствие данных желез приводит к судорогам вследствие гипокальциемии. Появление судорог у младенца в течение 48 часов после рождения свидетельствует о том, что у ребенка врожденная аплазия тимуса и дефицит Т-клеток. С данным синдромом связаны некоторые другие пороки развития, в том числе расщелина нёба (волчья пасть), щели в носу, низкопосаженные уши и неправильное формирование крупных кровеносных сосудов и сердца. Дети с пороками развития сердечно-сосудистой системы, как правило, умирают. Одно время считали, что этот синдром вызван нарушением нормального эмбрионального развития, которое не обусловлено наследственностью, поскольку не наблюдается семейной предрасположенности к заболеванию и оба пола поражаются одинаково. Впоследствии, однако, было установлено, что многие случаи заболевания связаны со структурными дефектами в 22-й хромосоме [9].
У детей, страдающих синдромом Ди Джорджи, либо совсем нет тимуса, либо он недоразвит, что встречается чаще. Такие дети чрезвычайно восприимчивы к вирусным, бактериальным и грибковым инфекциям и склонны к развитию таких опасных для жизни заболеваний, как цитомегаловирусная инфекция. Иногда в крови ребенка присутствует некоторое количество Т-клеток; такие дети способны сопротивляться инфекциям, при этом с возрастом число Т-клеток постепенно увеличивается [11].
Одна из форм нарушения иммунитета. Проявляется с рождения. Основные симптомы — тетания, обусловленная гипопаратиреозом; повышенная склонность к инфекциям, особенно вирусным и грибковым; аномалии развития костной системы, полости рта, сосудов, внутренних органов. Из инфекционных болезней чаще возникают бронхиты, пневмонии, диарея. Дети отстают и физическом развитии. Болезнь прогрессирует и без лечения к двум годам наступает летальный исход. При исследовании крови определяется низкий уровень, содержании кальции и высокий уровень фосфора, указывающие на паратиреендную недостаточность. Число лимфоцитов в периферической крови обычно нормальное, уровень иммуноглобулинов основных классов также в пределах нормы, но специфический иммунный ответ на введенный антиген снижен. Снижены реакции клеточного типа. Не развиваются и ослаблены реакции замедленной ин нечувствительности. Стимулированный лимфоузел обнаруживает избыточное число фолликулов и плазматических клеток. Наблюдаются уменьшение числа лимфоцитов в паракортикальных тимусзависимых зонах и белой пульпе селезенки, аплазия тимуса, гипоплазия паратиреоидных желез или аплазия их. Аплазия тимуса ведет к нарушению синтеза полноценных иммуноглобулинов. [12].
Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов. Характерны нормальные или повышенные уровни антител [9].
Показатели гуморального иммунитета у больных с синдромом Ди Джорджа полностью нормальны. Концентрация иммуноглобулинов всех классов нормальная. У некоторых больных повышена концентрация lgE, что, возможно, связано с отсутствием Т-супрессоров. У части больных не удается получить антитела в ответ на иммунизацию [11].
1. Обязательные лабораторные исследования:
- Анализ крови клинический4
- Тромбоциты 2
- Определение группы крови и резус фактора1
- Анализ мочи2
- Определение уровня кальция в крови2
- Бактериологические исследования содержимого из очагов с определением чувствительности к антибиотикам1